Oligonucleotides, such as antisense oligonucleotides, small interfering RNA (siRNA), aptamers, etc., can achieve the purpose of treating diseases by targeting pathogenic RNA or proteins and inhibiting their activity. However, many types of oligonucleotides have shortcomings such as poor active targeting, instability in vivo, and poor transmembrane ability. Therefore, researchers usually improve the functions and properties of nucleic acid molecules based on traditional oligonucleotides by covalent conjugation with other functional molecules, such as cell uptake, tissue delivery efficiency, bioavailability, catalytic effect, etc., to enhance the therapeutic effect of nucleic acids.
This review discusses the latest research progress on seven types of typical oligonucleotide conjugates in two development directions: delivery and catalytic degradation, aiming to provide ideas and references for the structural design and further clinical application of oligonucleotide conjugates.
In the past few decades, the field of nucleic acid drugs has undergone rapid development. Especially, antisense oligonucleotides (ASOs), small interfering RNA (siRNA), aptamers, and mRNA vaccines have attracted wide attention. Compared to traditional drugs, nucleic acid drugs have many advantages, such as high specificity, efficiency, and persistence.
In addition, nucleic acid drugs can act on non-druggable targets and can be used for post-transcriptional gene silencing or activation therapy. Therefore, nucleic acid drugs are considered very promising in biomedical research. As of now, about a dozen oligonucleotide-based drugs have been approved by the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMA).
However, the development of oligonucleotide drugs has not been smooth, and achieving the widespread use of oligonucleotide drugs in clinical settings remains highly challenging. For example, oligonucleotides are unstable in blood, have a short half-life, poor active targeting, and carry a negative charge that repels the cell membrane, leading to poor endocytosis and endosomal escape. To overcome these limitations and improve the delivery efficiency and clinical potential of oligonucleotides, oligonucleotides can be conjugated with other functional molecules to improve cell uptake, tissue delivery, bioavailability, and catalytic properties, thus enhancing the overall efficiency of oligonucleotides.
Currently, conjugated functional molecules include N-acetylgalactosamine (GalNAc), lipophilic molecules, peptides, small-molecule drugs, and antibodies, among others. Conjugating oligonucleotide drugs provides "navigation" to deliver oligonucleotides to specific cells and tissues, significantly reducing drug toxicity and becoming a crucial development direction for many pharmaceutical companies' next-generation delivery systems. Besides the development of delivery systems, oligonucleotide conjugates are also being developed for catalytic degradation of targets, such as oligonucleotide-peptide conjugates with catalytic functions. These conjugates can specifically degrade target RNA in the cytoplasm through sequence-specific recognition and peptide-catalyzed cleavage, thus inactivating the RNA.
Furthermore, new types of oligonucleotide conjugates, such as ribonuclease-targeting chimaeras (RIBOTACs), consist of small molecules targeting RNA with specific 3D structures and 2'-5' oligoadenylate (2'-5'A) recruiting ribonuclease L (RNase L), providing new strategies for targeting functional RNA with secondary structures. This review discusses the research progress of oligonucleotide conjugates from the development perspectives of delivery and catalytic degradation, focusing on some typical examples of oligonucleotide conjugates in recent years and discussing their mechanisms of action and potential applications in drug development.
Given the challenges of oligonucleotide transmembrane difficulty and poor active targeting, targeted delivery systems are a crucial focus. Mainstream oligonucleotide delivery systems include lipid nanoparticles (LNPs), oligonucleotide-ligand conjugates, naturally occurring cell delivery substances exosomes, and polymer-based siRNA delivery. Among these, oligonucleotide-ligand conjugates are considered highly promising for overcoming in vivo delivery bottlenecks, mainly relying on the characteristics of targeting ligand molecules.
The most representative GalNAc-siRNA conjugates have been validated, but they cannot solve drug targeting delivery outside the liver and have dosage and time dependency issues. Additionally, antibody drugs have shown explosive development in recent years. Utilizing the specificity of antibodies and antigens, new delivery methods such as oligonucleotide-antibody conjugates for delivering oligonucleotides have garnered wide attention and become effective means for specific delivery to tissues outside the liver.
Similar to oligonucleotide-antibody conjugate drugs, aptamer-drug conjugates are often used for studying cancer targeted therapy and immune mediated therapy. Moreover, research is ongoing on various oligonucleotide-targeting molecules, such as peptides and lipophilic small molecules.
The target sites of therapeutic oligonucleotides are within cells. Oligonucleotides have relatively large molecular masses and usually carry negative charges, preventing them from freely diffusing through cell membranes like lipophilic small molecule drugs. One primary challenge in developing oligonucleotide drugs is delivering them to diseased cells. Each liver cell has about 500,000 asialoglycoprotein receptors (ASGPRs), which are high-capacity and rapidly internalizing receptors.
GalNAc, a ligand for ASGPR, can quickly deliver GalNAc into the cell after binding to ASGPR. When GalNAc is covalently linked to oligonucleotides, the GalNAc portion can bind to ASGPR, targeting the delivery of oligonucleotides to liver cells. This method has become a breakthrough in the field of therapeutic oligonucleotides (see Figure 1).
Oligonucleotide-GalNAc conjugates have significant advantages in liver cell targeting . GalNAc conjugation increases oligonucleotide levels in liver cells by 6-7 times at the same dose, and this targeting effect has been proven well in clinical applications. The dosage required for gene knockout using oligonucleotide-GalNAc conjugates is 1/30-1/10 of unmodified oligonucleotides, thus small doses of oligonucleotide-GalNAc conjugates can also achieve good therapeutic effects.
siRNA-GalNAc conjugates have been widely used to treat liver-related diseases, with several drugs clinically approved, such as Alnylam Pharmaceuticals' givosiran, lumasiran, inclisiran, and vutrisiran for acute hepatic porphyria, primary hyperoxaluria type 1, hypercholesterolemia, and amyloidosis TTR-mediated amyloidosis, respectively. Overall, the advent of oligonucleotide-GalNAc conjugates has truly changed the landscape of the oligonucleotide therapeutic field.
Antibodies have been proven to be ideal functional molecules for drug delivery due to their high specificity, long half-life, and low immunogenicity. Antibody-drug conjugates (ADC) can selectively deliver chemotherapeutic drugs to cancer cells, improving therapeutic indices for many antibodies and expanding the potential for many small molecule drug therapies, becoming important new therapeutic methods.
Although ADC specificity is achieved through antibody specificity, toxicity reactions are still observable in clinical settings. Using more precise payload combinations may yield better-selective and safer ADCs. Oligonucleotides are such precise payloads, which inhibit protein production by introducing specific nucleic acid interference fragments, such as siRNA and ASOs.

Although oligonucleotides have selectivity, they also face issues like poor serum stability, low membrane permeability, and lack of tissue selectivity. Antibodies, with long half-lives, high selectivity, and targeting properties, are ideal partners for the targeted delivery of oligonucleotides. Antibody-oligonucleotide conjugates (AOC) combine siRNA and ASO precision with antibodies' delivery capabilities, synergizing the advantages of these two functional molecules.
The AOC field initially developed as a means to create strong diagnostic tools but has recently evolved into targeted treatment methods for various diseases (see Table 1). As the oligonucleotide field matures and selective tissue delivery becomes a crucial clinical application challenge, AOC has developed into potential single-agent therapies, achieving significant growth in recent years.
There are four common methods for preparing AOCs from oligonucleotides and antibodies (see Figure 2). These methods involve electrostatic interactions, affinity binding, direct conjugation, and using double-strands as the conjugation moiety.
The negative charge of the oligonucleotide backbone can tightly bind oligonucleotide and protein through electrostatic interactions with the positive charge of cationic portions in the antibody (such as protamine or polyarginine). This method does not require any chemical modifications and is suitable for various types of oligonucleotides. In a similar method, affinity binding between biotin-labelled oligonucleotides and avidin protein has successfully produced AOCs.
Although this is an effective and practical conjugation method, it still involves chemically modifying antibodies and oligonucleotides with biotin or streptavidin, usually requiring as many steps as other conjugation methods. Direct conjugation methods are more similar to antibody-drug conjugation methods, adding clickable groups to oligonucleotides and directly conjugating oligonucleotides to lysine, cysteine, or other engineered amino acids in antibodies. This method allows the use of the same linkers found in ADCs, such as cleavable disulfide linkers and other non-cleavable linkers.
Common direct conjugation methods use dibenzoazacyclooctyne (DBCO) and azide click chemistry reactions or strain-promoted azide-alkyne cycloaddition (SPAAC) reactions to produce conjugates. Double-stranded AOCs can be obtained through double-strand hybridization, where single-strand oligonucleotides are first conjugated to antibodies, and complementary strands hybridize to form double-stranded AOCs. Hybridization rates and specificities allow for constructing precise structures and combining them with chemotherapeutic drugs.

Aptamers are short single-stranded oligonucleotides with low toxicity and low immunogenicity. Their relative molecular mass and size are about 1/25 of antibodies, giving them better tissue penetration capabilities. In addition, aptamers can be screened using systematic evolution of ligands by exponential enrichment (SELEX) technology to obtain oligonucleotide sequences that can bind to various types of targets with high affinity and specificity.
Based on this, researchers proposed aptamer-drug conjugates (ApDC) similar to ADC for targeted cancer therapy. Typical ApDC consists of an aptamer, drug, and linker. ApDC can specifically deliver drugs to cancer cells, improving therapeutic specificity, enhancing efficacy, and reducing chemotherapy drug toxicity.
Combining ApDC with nanotechnology may increase drug accumulation at tumour sites, further enhancing ApDC's application potential. Tran et al. integrated floxuridine (FUdR) into an anti-nucleolin aptamer (AS1411) to obtain F-AS1411 and produced ApDC nanoparticles through rolling circle amplification polymerization. This conjugate showed enhanced tumour-targeting accumulation and significantly inhibited tumour growth.
siRNA, with its highly efficient sequence-specific gene silencing ability and unlimited target range, can also serve as the drug portion of ApDC. Various aptamer-mediated siRNA delivery systems have been used to enhance siRNA efficacy through targeted delivery. Zhang et al. developed a PROTACs-based aptamer-drug conjugate (ZL216), including the aptamer AS1411 as the targeting ligand and the small molecule ligand AHPC for E3 ligase VHL (see Figure 3A).
Based on differential expression of nucleolin on cell surfaces, ZL216 can specifically target breast cancer cells, induce nucleolin degradation in breast cancer cells, inhibit their proliferation and migration. Miao et al. constructed a bispecific aptamer chimera by simultaneously linking two different membrane protein aptamers to the cell surface insulin-like growth factor II receptor (IGFIIR) and membrane protein (see Figure 3B). The developed chimaera could transport membrane proteins to lysosomes for degradation via the lysosomal degradation pathway, achieving accurate degradation of epithelial-to-mesenchymal transition factor and receptor tyrosine kinase 7 membrane proteins.

Although aptamers have many advantages over antibodies, such as higher targeting binding affinity, ApDC's susceptibility to nucleases results in a short in vivo half-life, and non-specific protein adsorption leads to suboptimal aptamer-target protein binding effects, limiting the extensive in vivo application of aptamer-based drugs.
Cholesterol-modified siRNA is a widely studied lipophilic molecule conjugate designed to improve oligonucleotide pharmacokinetic properties in various tissues and enhance efficacy. Cholesterol, as one of the earliest reported lipophilic molecules, enhances oligonucleotide interaction with cell membranes, making it easier for oligonucleotides to be taken up by cells and regulate gene expression without using transfection agents.
Research shows a keen interest in discovering new hydrophobic molecules to enhance cell uptake of siRNA. Amino-modified siRNA can be conjugated with various small carboxyl lipids (such as cholesterol, fatty acids, and bile acids) to form lipid-siRNA complexes capable of interacting with lipoproteins (see Figure 4). These complexes can be efficiently delivered to the liver, intestines, and kidneys through specific lipoprotein-mediated receptors after interacting with lipoproteins. Cholesterol has been used to conjugate single-stranded short RNA molecules successfully for miRNA silencing.
Furthermore, research finds that G-quadruplex structures formed by oligonucleotide-lipid conjugates can increase their affinity for viral membrane proteins, thereby inhibiting viral entry into cells.

Recent studies have explored the application of oligonucleotide-lipid conjugates in transfecting cells and tissues outside the liver. Due to the unique nature of the blood-brain barrier, transfecting siRNA into primary neurons is very challenging. However, Alterman et al. found that cholesterol-tetraethylene glycol-functionalized siRNA could be effectively internalized in primary cortical neurons and successfully induce huntingtin gene silencing when injected into the mouse brain, effectively producing silencing effects.
Professor Xin Jing Tang's research team successfully developed Triton X-100 modified polyethyleneimine transfection vectors. Triton X-100, composed of hydrophobic head groups and hydrophilic PEG terminal long chains, optimizes the function of membrane proteins and significantly enhances eukaryotic cell membrane permeability for keratinocyte transfection and transdermal drug delivery.
This study utilized this vector to encapsulate Gas6 plasmids for gene therapy of androgenetic alopecia mice, demonstrating Triton X-100's application potential in hair follicle development and growth, presenting new strategies for gene therapy of skin-related diseases. Another characteristic of lipid-siRNA conjugates is that the lipid can promote siRNA entry into cells, utilizing this property to prepare nanoparticles capable of delivering therapeutic siRNA.
Professor Xin Jing Tang's research team developed a series of amphiphilic conjugates using natural components spermidine and vitamin E for siRNA delivery. They designed and synthesized a series of VESCs with different linkers, which self-assemble into VESC/siRNA nanocomplexes (see Figure 5); these siRNA nanocomplexes can be internalized through caveolae/lipid raft-mediated endocytosis, avoiding lysosomal capture, thereby achieving effective silencing of two endogenous target genes.

Conjugating peptides with therapeutic oligonucleotides can endow oligonucleotides with several properties, such as tissue and cell targeting, cell penetration, antiviral and antibacterial properties, and catalytic degradation properties.
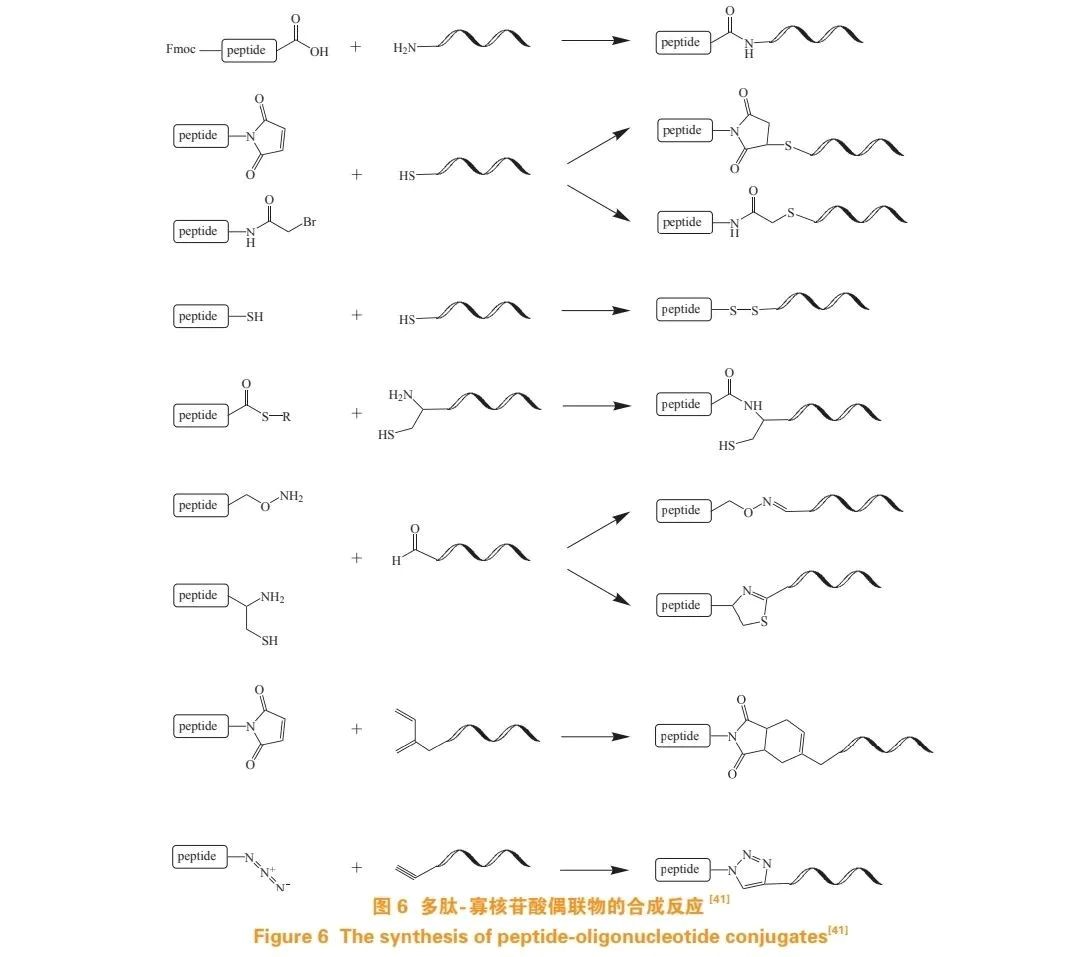
Two common methods for synthesizing peptide-oligonucleotide conjugates (POC) are:
Sequentially solid-phase synthesizing peptide and oligonucleotide fragments on the same solid-phase carrier, with the sequence order reversible;
Combining separately assembled peptide and oligonucleotide fragments on a solid phase and then conjugating them in the liquid phase.
The key to the second method is introducing a pair of mutually reactive chemical groups at appropriate positions in the peptide and oligonucleotide fragments, then connecting them through chemical selective conjugation reactions in solution or on a solid phase to form covalent bonds. Figure 6 presents common synthesis methods, including the reaction of Fmoc-protected peptides with amino-modified oligonucleotides, thiol-maleimide and thiol-bromoacetamide reactions, disulfide linkage, native ligation, oxime or thiazolidine reactions, Diels-Alder reaction, and copper-catalyzed azide-alkyne click reaction.
Among these, disulfide bonds and thiol-maleimide bonds are commonly used for conjugating oligonucleotides with cell-penetrating peptides (CPPs). A typical example is the conjugation of antisense oligonucleotides (ASOs) targeting inhibitors of differentiation 1 (Id1) with the F3-peptide fragment of tumour endothelial cells, which can inhibit tumour growth and metastasis in vivo. Additionally, ASOs conjugated with glucagon-like peptide-1 receptor (GLP1R) peptide ligands can inhibit GLP1R expression in pancreatic insulin-secreting β-cells, successfully downregulating the target gene within pancreatic β-cells.
CPPs are cationic or amphiphilic peptide fragments composed of 5 to 40 amino acid residues. CPPs can cross cell membranes and facilitate the intracellular transport of various active payloads, allowing therapeutic molecules to penetrate the cell membrane. CPPs are mainly classified into the following categories:
Cationic peptides, including Arg-rich peptides, Tat peptides, and penetration.
Amphiphilic peptides, such as transportan and poly-antigen peptides.
Hydrophobic peptides, such as C105Y and Pept1.
Mitochondrial micro peptides.
CPP-oligonucleotide conjugates are among the most extensively studied conjugates. A common conjugation strategy involves linking oligonucleotides with peptides that can recognize membrane receptors. For example, Arg-Gly-Asp (RGD) peptides, which have an affinity for integrins, or octreotide derivatives, which bind to somatostatin receptors, are conjugated with oligonucleotides to facilitate their cellular uptake.
Since cationic CPPs have more difficulty forming covalent bonds with negatively charged oligonucleotides, they are primarily used to enhance the uptake of charge-neutral phosphorodiamidate morpholino oligomers (PMOs) and peptide nucleic acids (PNAs) into cells.
Another class of peptides, known as homing peptides, has been developed through phage display technology. These peptides have been successfully used to deliver ASOs to cardiac tissue or to transport siRNA and DNA plasmids to the spinal cord or microglial cells. Additionally, certain peptides have been applied in the treatment of Duchenne muscular dystrophy (DMD), such as enhanced delivery oligonucleotide (EDO) (PepGen) and SRP-5051 (Sarepta).
The conjugation of targeting molecules with catalytic molecules enables targeted catalytic activity. For instance, oligonucleotide-conjugated catalytic peptides leverage the sequence-specific recognition of oligonucleotides while relying on the direct catalytic cleavage function of peptides. This approach allows for the selective degradation of complex RNA structures, rendering them inactive.
Such catalytic conjugates, inspired by the targeted protein degradation mechanism, have led to the development of oligonucleotide-based conjugates, with RIBOTACs being a typical example. RIBOTACs utilize a small molecule as a ligand to target specific RNA structures, while the conjugated oligonucleotide recruits the catalytic enzyme RNase L to degrade the target RNA. This strategy presents a novel therapeutic approach for targeting RNA.
Site-selective artificial ribonucleases (ss-aRNases) are conjugates of oligonucleotides and catalytic domains. They inherently combine the RNA sequence recognition capability of oligonucleotides with the catalytic domain's ability to cleave phosphodiester bonds.
Generally, aRNases mimic natural enzymes to some extent, such as the catalytic centre of ribonuclease A (RNase A), which contains functional groups such as imidazole, guanidine, and amine residues (e.g., histidine, arginine, or lysine). Several peptide-based RNases have demonstrated activity in early studies. Notably, peptides with alternating hydrophobic and basic amino acids (typically containing 20 or more residues) exhibit RNase activity, with the most active being those composed of alternating leucine and arginine residues.
The [(LeuArg)₂Gly]₂ peptide, which contains alternating leucine and arginine residues, has been identified as an effective catalytic peptide. When conjugated with an oligonucleotide, it forms an RNase. Studies have shown that this conjugate can successfully downregulate overexpressed pathogenic miRNAs, effectively and selectively inhibiting disease-associated RNA both in vitro and in vivo. One notable application is its ability to selectively degrade the oncogenic miR-21, thereby inducing cancer cell apoptosis, inhibiting cell invasion and proliferation, and suppressing tumour growth in vivo, demonstrating significant therapeutic potential.
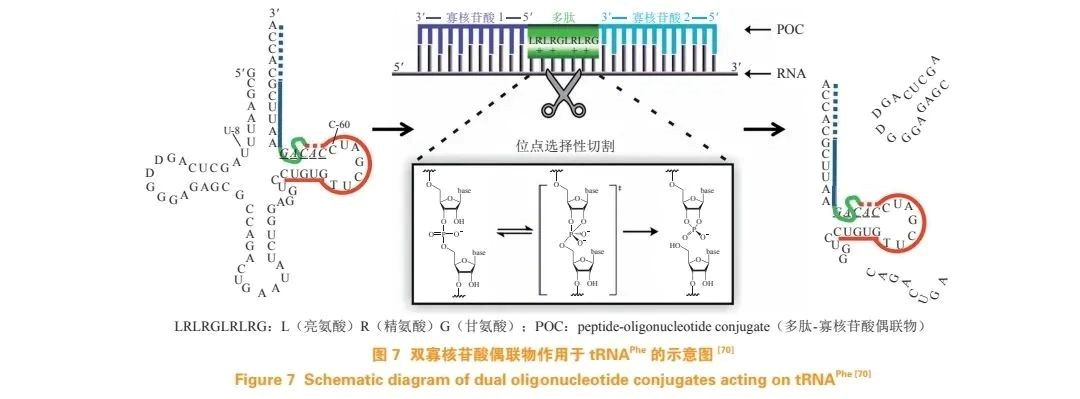
Staroseletz et al. designed and synthesized a new peptide-oligonucleotide conjugate (POC), where the catalytic peptide was placed between two oligonucleotide recognition sequences (see Figure 7). Although tRNAPhe is not a clinically relevant RNA target, it contains secondary structural elements found in many mRNAs, miRNAs, and viral RNAs, making it a useful conceptual model target for developing these novel POCs. Their study demonstrated that under physiological conditions, POCs could efficiently cleave tRNAPhe, and they successfully screened for POCs with optimal RNA-cleaving activity.
Many RNA targets exhibit unique structural features that regulate various RNA processing mechanisms, making them valuable therapeutic targets. Compared to antisense oligonucleotide (ASO)-based therapies, small molecules are often better suited for binding structured RNA, making them more effective for targeting highly structured RNA molecules.
The design of small molecules targeting three-dimensional RNA structures has been a rapidly evolving field. Several methods enable the prediction and synthesis of small molecules that selectively bind disease-associated RNA functional sites. As RNA secondary structures fold further, they form tertiary structures, which allow small molecules to bind to unique structural motifs, thereby modulating transcript functions.
Numerous structured RNAs have been successfully targeted by small molecules. For instance, researchers have developed compounds that bind to expanded repeat sequences in viral RNA, which can mitigate disease phenotypes associated with myotonic dystrophy (DM) types 1 and 2 and amyotrophic lateral sclerosis (ALS). Another example is small molecules targeting HIV reverse transcription, which significantly impact HIV infection progression.
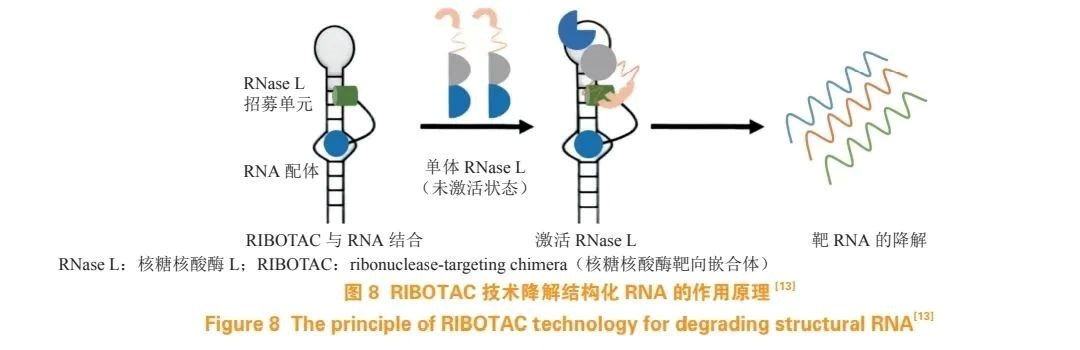
Tong et al. developed RIBOTACs by conjugating a small molecule ligand with an RNase-recruiting unit, thereby recruiting RNases to specific transcripts and promoting their degradation. These RNAs play critical roles in gene regulation, making RIBOTACs a promising approach for treating cancer, viral infections, and other diseases. Both in vitro and in vivo studies have demonstrated the broad potential of RIBOTACs, but further research is needed to fully understand their capabilities and limitations.
RIBOTACs are chimeric molecules composed of two functional parts:
A targeting unit that directs RNases to the desired RNA target.
A recruiter unit that brings in and activates an RNase, leading to RNA cleavage and degradation (see Figure 8).
RNase L cleaves RNA in a sequence-specific manner, targeting both single-stranded and double-stranded RNA within cells. Human RNase L consists of 735 amino acid residues and requires activation by 2'-5'A binding. When nuclease enzymes are recruited by RIBOTACs, their proximity to the target RNA enables efficient cleavage. After the target RNA is degraded, RIBOTACs are released and can act on additional RNA targets, allowing for selective, catalytic, and sub-stoichiometric elimination of toxic RNA.
RIBOTACs offer a new approach for both RNA research and therapeutic development for RNA-related diseases. Beyond RIBOTACs, various RNA degradation strategies have emerged, including:
PROTACs (proteolysis-targeting chimeras),
LYTACs (lysosome-targeting chimeras),
MADTACs (macroautophagy degradation-targeting chimeras).
These emerging technologies provide innovative strategies for drug discovery.
Oligonucleotide conjugation technology is an innovative drug design strategy that expands therapeutic applications, enhances drug efficacy, and reduces adverse effects. This review summarizes recent research on oligonucleotide conjugates, demonstrating how conjugation with antibodies, small-molecule drugs, lipophilic molecules, or peptides improves the functionality and properties of nucleic acid molecules. These enhancements include better cellular uptake, improved tissue distribution, increased bioavailability, and catalytic effects.
However, oligonucleotide conjugates still face numerous challenges and unexplored areas. Key issues that require further investigation include:
How to overcome the biological instability and immunogenicity of oligonucleotides.
How to achieve precise targeting and controlled intracellular release.
How to reduce manufacturing costs and improve production efficiency.
Through continuous research and innovation of biotechnical solutions, oligonucleotide conjugates hold promise for personalized medicine and precision therapy, offering new opportunities for human health and therapeutic advancements.





